|
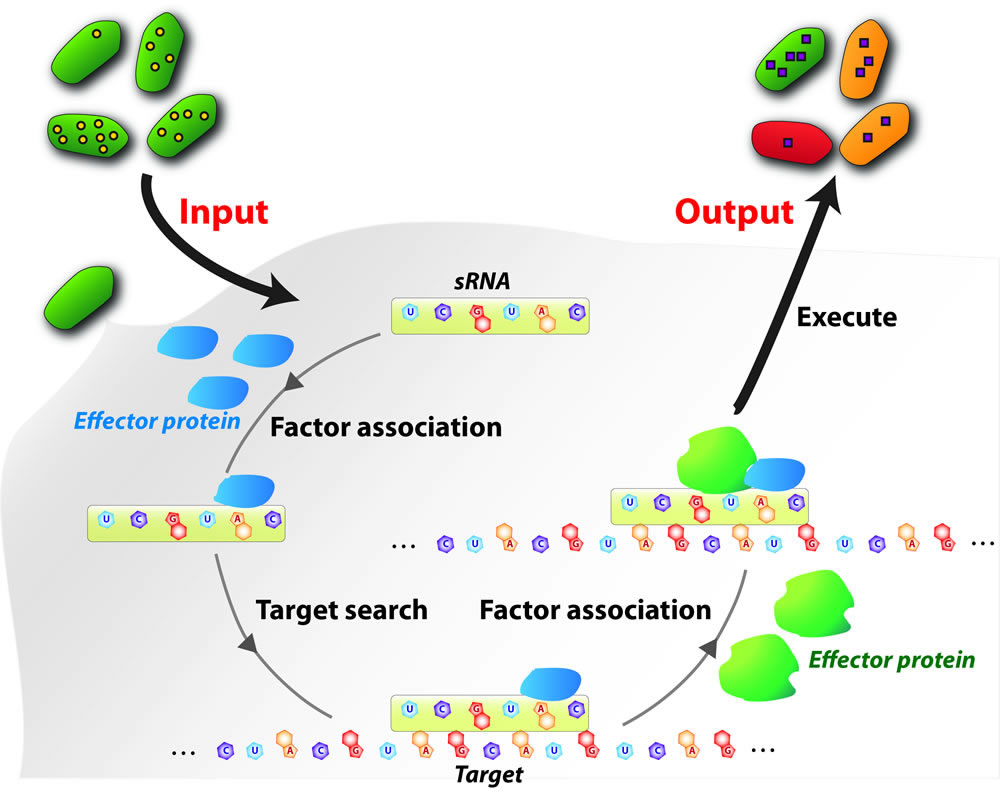 Small regulatory noncoding RNAs (here broadly
defined as sRNAs) play critical roles in regulating genes involved in
almost all cellular processes, including development, apoptosis, stress
responses, tumorigenesis, infection, and immunity. Due to their
specificity and versatility, sRNAs have inspired broad applications
including new therapies for human diseases (e.g., small hairpin RNAs)
and genome engineering (e.g., CRISPR/Cas systems), among other
applications. That said, a precise quantitative description of the
fundamental mechanism of sRNA-mediated regulation and interference can
largely benefit the further improvement of the efficiency and
robustness of these applications by providing critical models and
parameters. Our research aims at providing a quantitative description
at the molecular, cellular, and the systems levels.Using bacteria as
model systems,our missions are to understand the molecular mechanisms
by which sRNAsmodulate messenger RNA (mRNA) translation and
degradation, as well as physiological response caused by sRNA-mediated
regulation in the context of pathogenic bacteria-host interactions. Small regulatory noncoding RNAs (here broadly
defined as sRNAs) play critical roles in regulating genes involved in
almost all cellular processes, including development, apoptosis, stress
responses, tumorigenesis, infection, and immunity. Due to their
specificity and versatility, sRNAs have inspired broad applications
including new therapies for human diseases (e.g., small hairpin RNAs)
and genome engineering (e.g., CRISPR/Cas systems), among other
applications. That said, a precise quantitative description of the
fundamental mechanism of sRNA-mediated regulation and interference can
largely benefit the further improvement of the efficiency and
robustness of these applications by providing critical models and
parameters. Our research aims at providing a quantitative description
at the molecular, cellular, and the systems levels.Using bacteria as
model systems,our missions are to understand the molecular mechanisms
by which sRNAsmodulate messenger RNA (mRNA) translation and
degradation, as well as physiological response caused by sRNA-mediated
regulation in the context of pathogenic bacteria-host interactions.
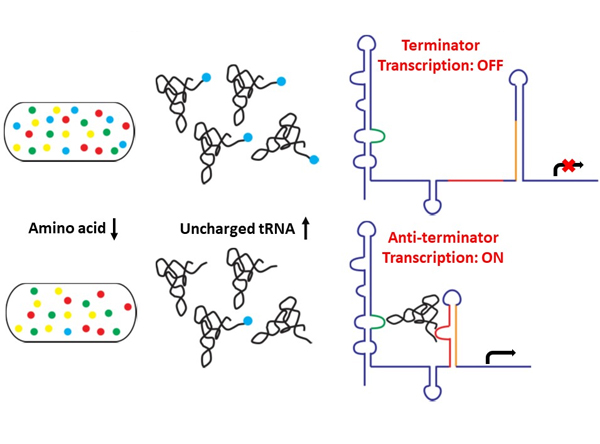 Riboswitches
are cis-regulatory RNA elements located upstream of messenger RNAs
(mRNA). Binding of ligands to the aptamer domain of the riboswitches
can lead to conformational changes in the expression platform of the
riboswitches, hereby affecting the transcription or translation of the
controlled mRNAs. T-box riboswitches are widely found in Gram-positive
bacteria, including pathogens, and are regulating essential genes,
including aminoacyl tRNA synthetases, and enzymes or components
involved in amino acid biosynthesis and transport. Therefore, T-box
riboswitches can potentially be a target for developing new
antibiotics. In addition, different from most small molecule binding
riboswitches, T-box riboswitches recognize tRNA molecules as ligand,
serving an excellent paradigm to understand RNA-based molecular
interactions. The regulation mechanism by the T-box riboswitch involves
transcription read-through or pre-mature transcriptional termination
depending on the aminoacylation status of the bound tRNA. Using
single-molecule FRET, we are investigating the binding dynamics of
T-box and its tRNA ligand. Riboswitches
are cis-regulatory RNA elements located upstream of messenger RNAs
(mRNA). Binding of ligands to the aptamer domain of the riboswitches
can lead to conformational changes in the expression platform of the
riboswitches, hereby affecting the transcription or translation of the
controlled mRNAs. T-box riboswitches are widely found in Gram-positive
bacteria, including pathogens, and are regulating essential genes,
including aminoacyl tRNA synthetases, and enzymes or components
involved in amino acid biosynthesis and transport. Therefore, T-box
riboswitches can potentially be a target for developing new
antibiotics. In addition, different from most small molecule binding
riboswitches, T-box riboswitches recognize tRNA molecules as ligand,
serving an excellent paradigm to understand RNA-based molecular
interactions. The regulation mechanism by the T-box riboswitch involves
transcription read-through or pre-mature transcriptional termination
depending on the aminoacylation status of the bound tRNA. Using
single-molecule FRET, we are investigating the binding dynamics of
T-box and its tRNA ligand.
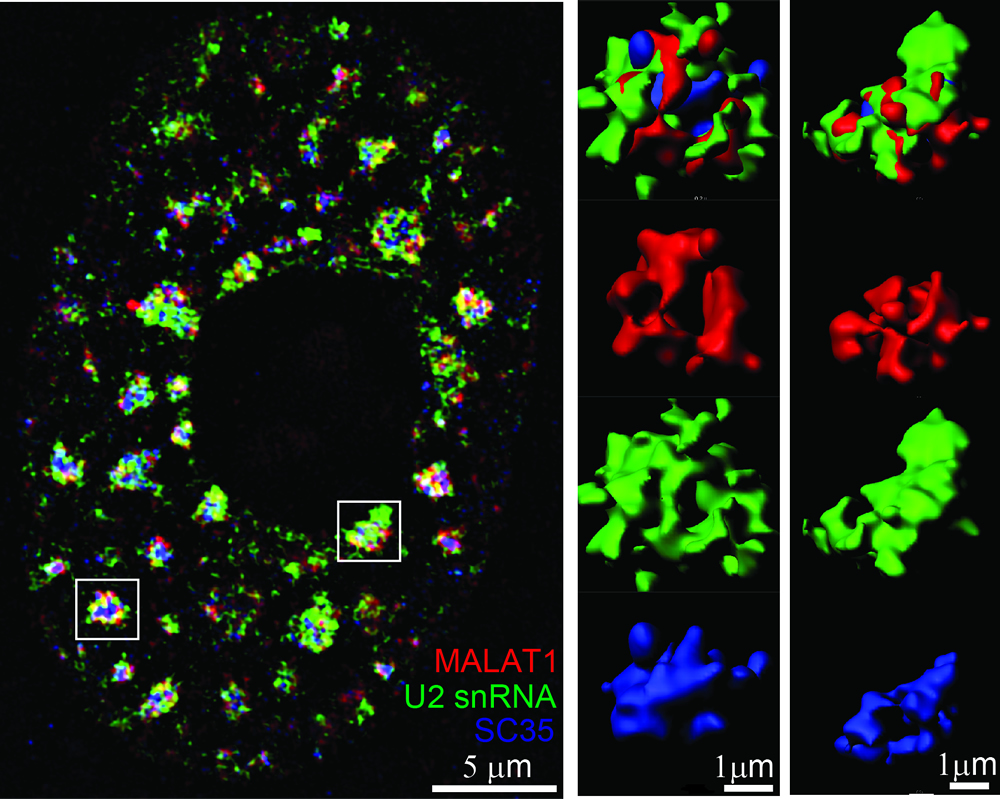 Eukaryotic cells are
significantly compartmentalized. Dynamic RNA localization to these
subcellular compartments profoundly impacts gene expression and other
vital
cellular activities, and can provide a novel way for stress response
and
adaptation. In particular, multivalent
interactions among certain RNA
and protein species can drive the formation of membraneless condensates. Nuclear speckles
represent one type of such membraneless bodies in the nucleus of higher
eukaryotes,
enriched in snRNP species, splicing factors, polyadenylated RNAs (polyA
RNAs) and certain long noncoding
RNAs (lncRNAs).
Formation of nuclear speckle requires the scaffold proteins SON and
SRRM2. Eukaryotic pre-mRNA after
transcription undergoes a
series of processing steps to become mature mRNA, including 5’ capping,
splicing to remove introns and ligate exons, 3’ polyadenylation, and
export to
cytoplasm. Nuclear speckle are suggested to play roles in several mRNA processing
steps,
including transcription enhancement, splicing quality control and RNA
export; and have been implicated in neurodegenerative diseases, infectious diseases
and
cancers. We are currently investigating why certain RNAs are preferentially localized to nuclear speckles over
others, and
what are the functional impacts of speckle localization. Eukaryotic cells are
significantly compartmentalized. Dynamic RNA localization to these
subcellular compartments profoundly impacts gene expression and other
vital
cellular activities, and can provide a novel way for stress response
and
adaptation. In particular, multivalent
interactions among certain RNA
and protein species can drive the formation of membraneless condensates. Nuclear speckles
represent one type of such membraneless bodies in the nucleus of higher
eukaryotes,
enriched in snRNP species, splicing factors, polyadenylated RNAs (polyA
RNAs) and certain long noncoding
RNAs (lncRNAs).
Formation of nuclear speckle requires the scaffold proteins SON and
SRRM2. Eukaryotic pre-mRNA after
transcription undergoes a
series of processing steps to become mature mRNA, including 5’ capping,
splicing to remove introns and ligate exons, 3’ polyadenylation, and
export to
cytoplasm. Nuclear speckle are suggested to play roles in several mRNA processing
steps,
including transcription enhancement, splicing quality control and RNA
export; and have been implicated in neurodegenerative diseases, infectious diseases
and
cancers. We are currently investigating why certain RNAs are preferentially localized to nuclear speckles over
others, and
what are the functional impacts of speckle localization.
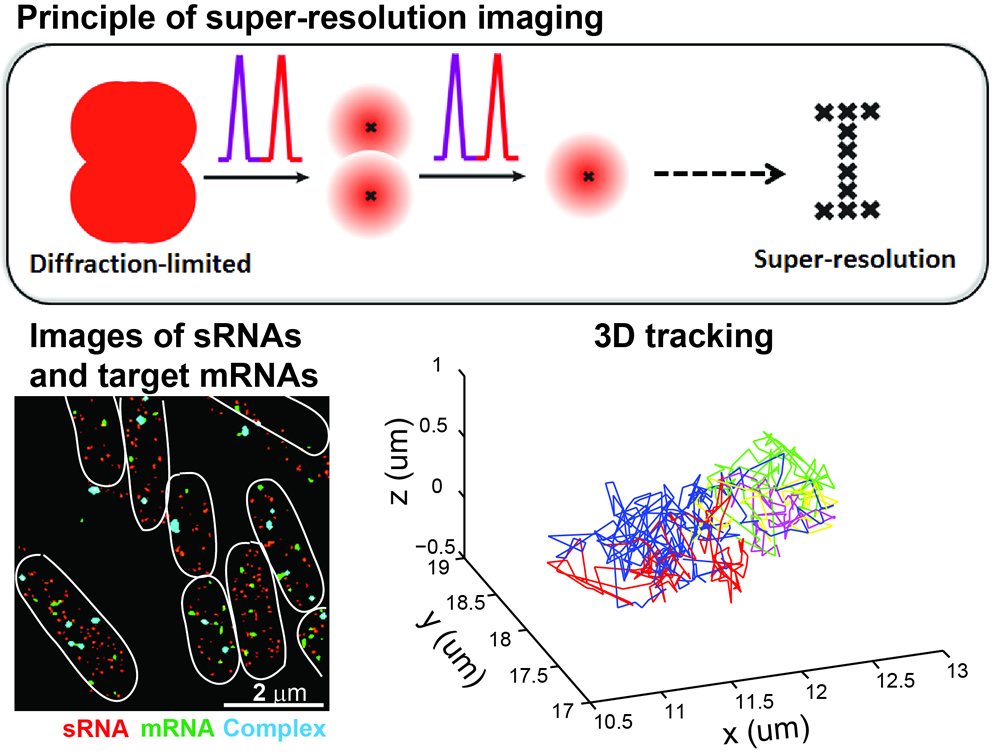 The
center of a single fluorophore can be very accurately determined in the
diffraction limited spot. Taking the advantage of photophysical
properties of certain fluorophores that can stochastically blink
between the bright and dark states, in single-molecule localization
based super-resolution imaging, only a small fraction of fluorophores
are activated and localized each time. By repeating the
reactivating-and-imaging cycle many times and combining many of such
frames together, a super-resolution image can be reconstructed that
usually has ~10-fold enhancement in the resolution compared to
diffraction-limited fluorescence microcopy. Super-resolution imaging
provides a powerful tool for investigating subcellular localization,
higher-order architectures of sub-compartments and inter-molecular
interactions as well as tracking the motions of individual molecules
inside the cell. The
center of a single fluorophore can be very accurately determined in the
diffraction limited spot. Taking the advantage of photophysical
properties of certain fluorophores that can stochastically blink
between the bright and dark states, in single-molecule localization
based super-resolution imaging, only a small fraction of fluorophores
are activated and localized each time. By repeating the
reactivating-and-imaging cycle many times and combining many of such
frames together, a super-resolution image can be reconstructed that
usually has ~10-fold enhancement in the resolution compared to
diffraction-limited fluorescence microcopy. Super-resolution imaging
provides a powerful tool for investigating subcellular localization,
higher-order architectures of sub-compartments and inter-molecular
interactions as well as tracking the motions of individual molecules
inside the cell.
Single-molecule
detection 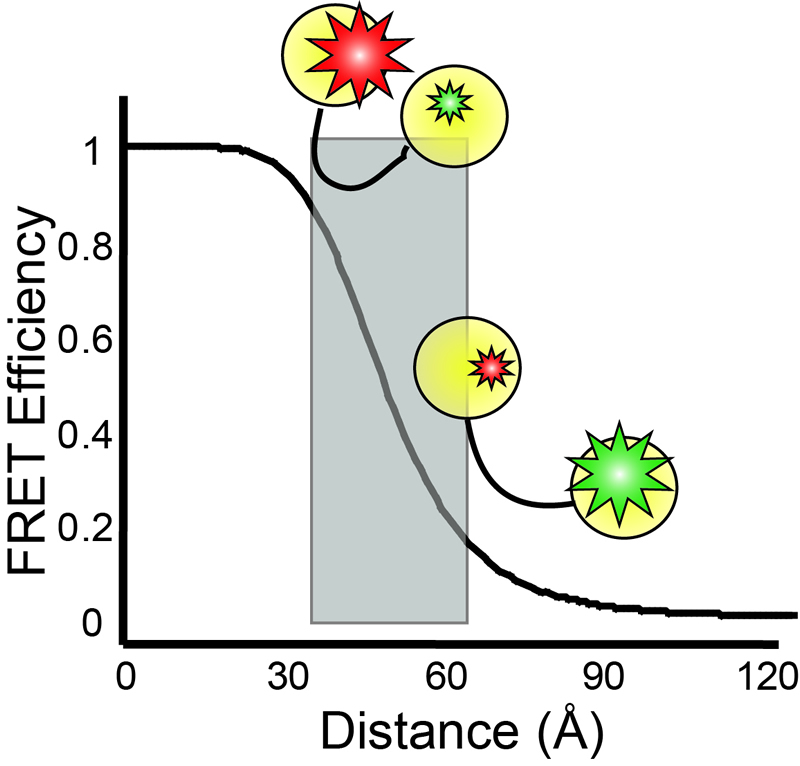 Single-molecule
fluorescence microscopies are widely nowadays in biophysical research.
By fluorescently labeling different molecules or multiple domains on
the same molecule, association/dissociation of factors, critical
conformational dynamics, and the temporal order of various events can
be simultaneously measured, and heterogeneities in the kinetic pathways
can be revealed.In particular, single-molecule fluorescence resonance
energy transfer (smFRET) is especially powerful for probing
conformational changesfor biomolecules, as the energy transfer
efficiency is inverse proportional to the sixth power of the distance
between a pair ofthe donor and the acceptor dyes, making FRET a very
sensitive microscopic ruler at nanometer scale. Single-molecule
fluorescence microscopies are widely nowadays in biophysical research.
By fluorescently labeling different molecules or multiple domains on
the same molecule, association/dissociation of factors, critical
conformational dynamics, and the temporal order of various events can
be simultaneously measured, and heterogeneities in the kinetic pathways
can be revealed.In particular, single-molecule fluorescence resonance
energy transfer (smFRET) is especially powerful for probing
conformational changesfor biomolecules, as the energy transfer
efficiency is inverse proportional to the sixth power of the distance
between a pair ofthe donor and the acceptor dyes, making FRET a very
sensitive microscopic ruler at nanometer scale.
 We have employed a density based clustering
analysis algorithm, DBSCAN, to analyze RNA copy numbers. Spots
corresponding to individual localization events in a reconstructed
super-resolution image are segregated into clusters based on their
spatial density. Clustered data are then superimposed to the DIC image
and the boundaries of individual cells were identified using MATLAB
code such that clusters are allocated into individual cells. After
clustering analysis and cluster allocation, information derived
includes: (1) total number of clusters in each cell, which approximates
the total number of RNAs in low copy number cases; (2) number of
localization spots in each cluster, which we use to build the
characteristic distribution of number of spots per RNA; (3) total
number of clustered spots in each cell, which is the product of (1) and
(2) and is used for estimating the copy number of RNA per cell; (4)
average radius of individual clusters; (5) center coordinates of
individual cells. We have employed a density based clustering
analysis algorithm, DBSCAN, to analyze RNA copy numbers. Spots
corresponding to individual localization events in a reconstructed
super-resolution image are segregated into clusters based on their
spatial density. Clustered data are then superimposed to the DIC image
and the boundaries of individual cells were identified using MATLAB
code such that clusters are allocated into individual cells. After
clustering analysis and cluster allocation, information derived
includes: (1) total number of clusters in each cell, which approximates
the total number of RNAs in low copy number cases; (2) number of
localization spots in each cluster, which we use to build the
characteristic distribution of number of spots per RNA; (3) total
number of clustered spots in each cell, which is the product of (1) and
(2) and is used for estimating the copy number of RNA per cell; (4)
average radius of individual clusters; (5) center coordinates of
individual cells.
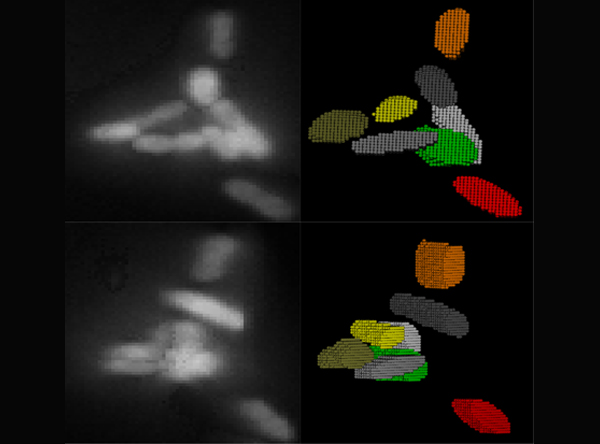 In
order to correctly correlate a genotype or phenotype to a specific cell
from our imaging experiments, images containing a population of cells
must first be properly segmented. We have developed an image analysis
package, Seg-3D, for the segmentation of bacterial cells in
three-dimensional (3D) images, based on local thresholding, shape
analysis, concavity-based cluster splitting, and morphology-based 3D
reconstruction. Seg-3D enables a proper segmentation with minimal user
input, even when cells are clustered or overlapping in three
dimensions. The reconstructed cell volumes allow us to directly
quantify the fluorescent signals from biomolecules of interest within
individual cells. Seg-3D is an efficient and simple program that can be
used to analyze a wide variety of single-cell images, especially for
biological systems involving random 3D orientation and clustering
behavior, such as bacterial infection or colonization. In
order to correctly correlate a genotype or phenotype to a specific cell
from our imaging experiments, images containing a population of cells
must first be properly segmented. We have developed an image analysis
package, Seg-3D, for the segmentation of bacterial cells in
three-dimensional (3D) images, based on local thresholding, shape
analysis, concavity-based cluster splitting, and morphology-based 3D
reconstruction. Seg-3D enables a proper segmentation with minimal user
input, even when cells are clustered or overlapping in three
dimensions. The reconstructed cell volumes allow us to directly
quantify the fluorescent signals from biomolecules of interest within
individual cells. Seg-3D is an efficient and simple program that can be
used to analyze a wide variety of single-cell images, especially for
biological systems involving random 3D orientation and clustering
behavior, such as bacterial infection or colonization.
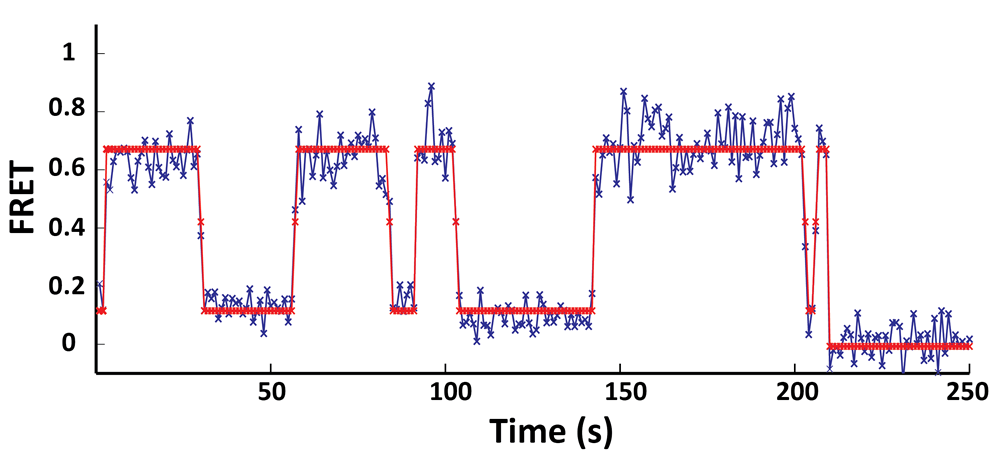 In
collaboration with Prof. Chris Wiggins group (Applied Physics and
Applied Mathematics, Columbia University), during my graduate research,
we have developed a variational Bayesian approach that allows us to
generalize the concept of maximum likelihood to determining the most
likely model (e.g. the number of conformational states in the
biological system) as well as the most likely parameters (e.g. the
transition rates between conformational states) that best describe the
single-molecule time trajectories. We have coded this algorithm into a
widely available opensource software package called vbFRET
(http://vbfret.sourceforge.net/). In
collaboration with Prof. Chris Wiggins group (Applied Physics and
Applied Mathematics, Columbia University), during my graduate research,
we have developed a variational Bayesian approach that allows us to
generalize the concept of maximum likelihood to determining the most
likely model (e.g. the number of conformational states in the
biological system) as well as the most likely parameters (e.g. the
transition rates between conformational states) that best describe the
single-molecule time trajectories. We have coded this algorithm into a
widely available opensource software package called vbFRET
(http://vbfret.sourceforge.net/).
|